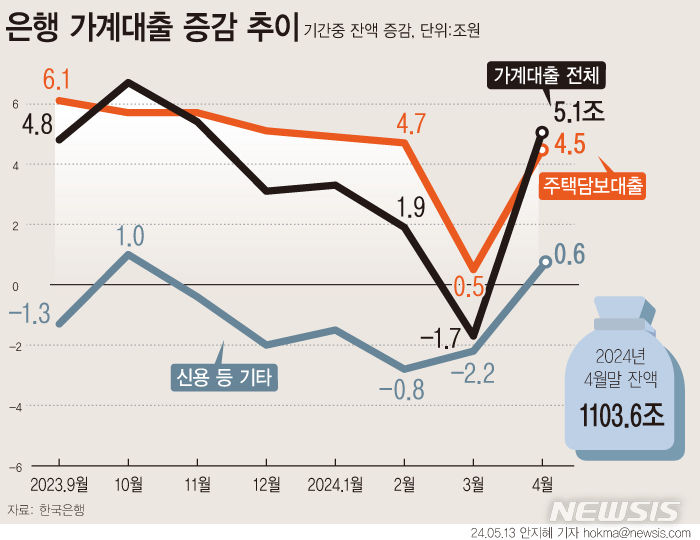








"의약품 글로벌 임상서 '인종 다양성' 필요"…美FDA 권고
등록 2023.11.09 14:05:35
인구집단 불균형, 약물 안전성·독성 파악 어려워

[서울=뉴시스]황재희 기자 = 글로벌 임상 추진 시 고려해야 하는 ‘인종 다양성’을 두고 미국 식품의약국(FDA)이 잇따라 가이던스를 제시하고 있어 주목된다.
9일 식품의약품안전평가원 ‘미국 의약품 규제 동향 브리프’ 자료에 따르면, FDA는 ‘임상시험 인구 다양성 확보’를 위해 작년에 이어 올해도 추가 가이던스를 내놨다.
FDA는 임상시험에서 인종 다양성을 확보하기 위해 2016년부터 가이던스 발표, 이해관계자 대상 홍보, 소수인종 대상 정보 제공을 위한 홈페이지 운영 등의 노력을 기울이고 있으나, 임상시험에서 여전히 과소 대표되는 집단이 존재하고 있다.
이에 FDA는 작년 ‘항암제 임상시험에 노인 포함’ 및 ‘임상시험에서 과소 대표되는 소수인종 및 소수 민족 피험자 등록을 개선하기 위한 계획’에 대한 가이던스에 이어 지난 8월 ‘환자의 다양성 확보를 위한 시판 후 임상 데이터 확보’에 대한 가이던스를 추가로 발표했다.
FDA는 제약사가 의약품 시판 후 과소 대표 인구집단 관련 데이터를 얻고자 한다면 제품 개발 초기부터 FDA와 협력해 인종 다양성 계획을 제출하는 것을 권고하고 있다. 만약 의도한 다양성 목표를 달성할 수 없는 경우, 그다음 전략도 FDA와 논의해야 한다.
FDA는 시판 후 데이터를 요청하기 위한 메커니즘으로 시판 후 요건(PMR)와 시판 후 약정(PMC) 두 가지 방법을 제시했다.
만약 부작용 보고가 불충분할 경우, FDA는 PMR에 따라 시판 후 연구 또는 임상시험 승인 후 연구를 제약사에 요구한다. 또 임상시험에서 특정 하위 집단이 과소 대표되는 경우, PMC 하에서 해당 하위 집단의 임상적 유익성 및 안전성을 특정하는 연구를 실시해야 한다.
제약사는 이때 단일군 임상시험, 무작위 임상시험, 실사용 데이터(RWD), 메타분석 등 다양한 자료 및 연구 방법을 활용할 수 있다.
기존 주요 분석에서 과소 대표됐던 하위 인구집단을 별도의 코호트로 등록해 분석하거나, 하위 인구집단에 따라 잠재적인 예후가 달라질 것으로 예상되는 경우 하위 집단에 층화한 연구 설계를 적용하면 된다. 만약 실제 데이터를 시판 후 연구 자료로 활용하고 싶다면 개발 초기 단계에서 FDA 검토부서와 이를 논의해야 한다.
임상시험에 포함되는 인종, 성별, 민족 등의 인구집단 불균형이 나타나면, 임상 데이터 및 치료 지표, 약물의 안전성·독성을 파악하는 데 한계점으로 작용한다. 과소 대표되는 인구집단에 대한 데이터 부재는 그 인구집단에 대한 지식 부재를 의미하는 것으로, 이 경우 의약품 및 백신의 안전성·효능을 확신할 수 없다.
실제로 한 연구에 따르면, 2014년부터 2019년까지 FDA 승인을 받은 신약 중 약 10%가 인종, 민족 또는 약물 유전학에 따라 노출 및 반응에 차이가 있었으며, 특정 약물의 경우는 인구집단별 처방 권장 사항을 별도로 작성해야 할 만큼 충분하게 큰 것으로 나타났다.
연구팀은 “우리나라도 여성, 소아, 임산부 등 임상시험에서 과소 대표될 수 있는 인구집단을 임상시험에 포함해 이들의 임상시험 데이터를 확보하는 활동을 강화할 필요가 있다”며 “또 인종 다양성이 향후 증가할 것으로 예상되는바, FDA 등 국외 동향을 참고해 장기적인 관점에서의 검토가 필요할 것”이라고 분석했다.
◎공감언론 뉴시스 [email protected]
Copyright © NEWSIS.COM, 무단 전재 및 재배포 금지